Improving Spectroscopic Understanding of Actinides
- Joesph M. Kasper

One longstanding challenge in actinide science is understanding the reactivity and chemical bonding of actinide complexes. These complexes, such as those involving uranium, occur both naturally in the environment as well as artificially, as a component of nuclear waste. Ideally, one could separate out the various elements to both isolate hazardous material and reclaim and recycle valuable rare earth elements. This requires a thorough understanding of how strongly the different metals bind to other ions or molecules, known as ligands. Experiments with actinides are, however, challenging due to their radiological properties; fortunately, we can use theoretical models and perform calculations using quantum mechanics to better understand experimental data as well as explore both real and hypothetical complexes.
The main goal of computational chemistry is to solve the Schrödinger or Dirac equation for a system of electrons in the potential generated by the atoms in the molecule. Doing so allows one to predict any of its observable properties. However, due to the complexity of the many-body problem, these equations cannot be precisely solved in practice; instead, approximations must be made, requiring a careful balance of computational cost and accuracy. Understanding the accuracy of different levels of calculation is essential for advancing a predictive capability for actinide bonding that can be used to inform the most promising future chemistries. In this work, calculations using different levels of theoretical approximations and computational cost were performed to interpret experimental X-ray absorption spectroscopy (XAS) data on actinide bonding.
Measuring chemical bonds with X-rays
X-ray absorption spectroscopy is an experimental technique that has gained prominence as a method to analyze the bonding and electronic structure of metal complexes. In XAS, high-energy photons excite the tightly bound core electrons in atoms or molecules to unoccupied quantum mechanical states, as depicted in Fig. 1 (see Advancing Understanding of Actinides in Aqueous Media). These levels are sensitive to bonding in the metal complex and its local environment such that small changes in the structure produce noticeably different spectra. With a theoretical framework or model, both the position and intensity of the peaks in the absorption spectrum can be interpreted and used to evaluate the strength and nature of chemical bonding.
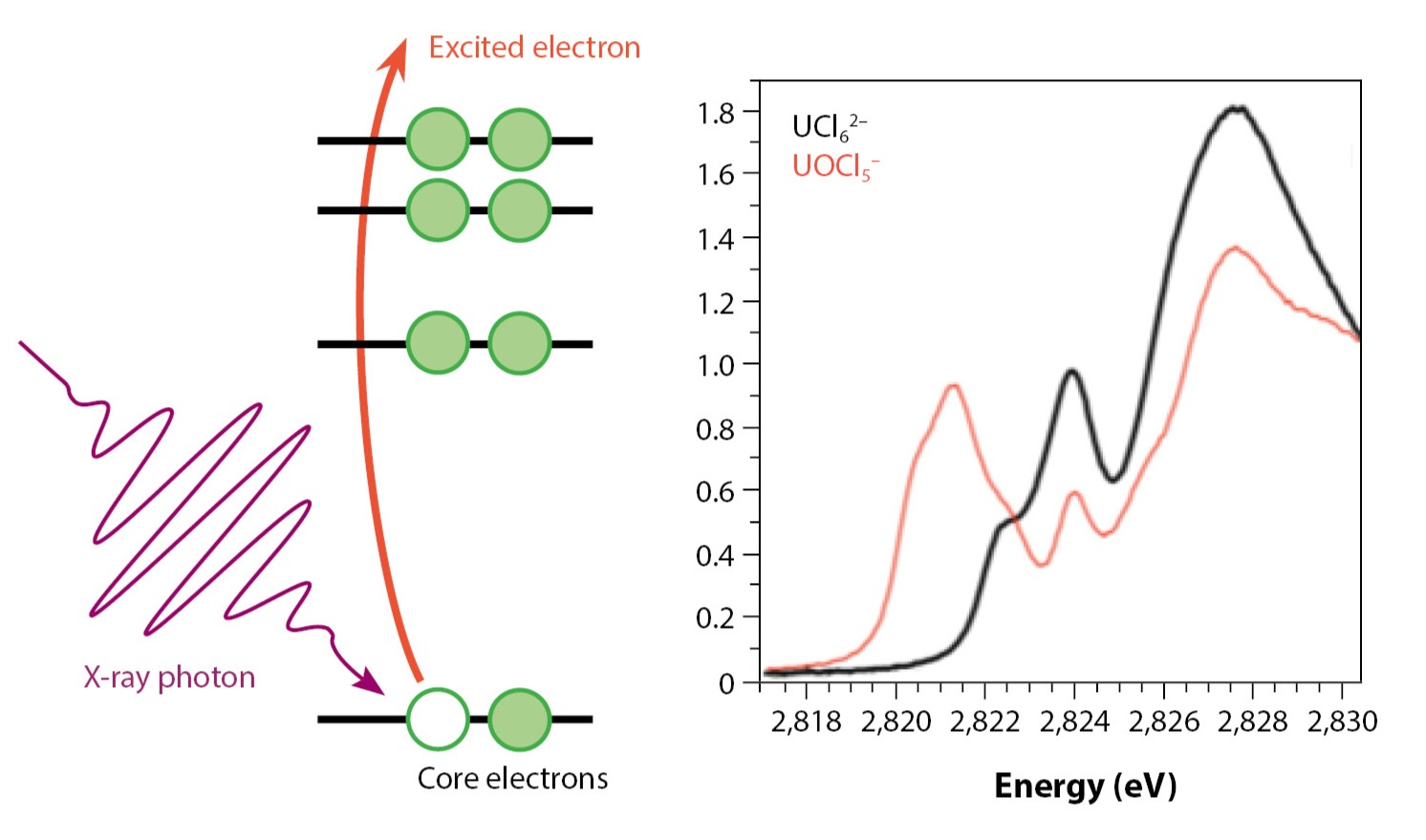
One of the reasons XAS is very useful for studying bonding is the strong energy dependence on the atomic number of the element. This means that it is relatively easy to pick out a particular metal or ligand atom of interest and resolve specific spectral lines or peaks instead of having many overlapping features. While the idea of measuring metal bonding by probing the ligand atoms was initially used within transition metal (d-block) chemistry, scientists at Los Alamos have applied this technique to actinide complexes and were able to show that much richer chemistry occurs with the elements than was previously understood. Unlike their lanthanide () counterparts, in which the electrons are well shielded from the valence electrons, in complexes with elements, these electrons are exposed and able to participate in bonding. Depending on the particular ligands, oxidation state, symmetry, and energetic match, the amount of covalency can be tuned. The variations in chemical bonding available with actinide species have renewed interest in exploring the chemical space of actinide complexes for many different applications.
Advanced relativistic quantum chemistry calculations
For high-Z elements such as the actinides, ab initio calculations are complicated due to both a large number of electrons and the need to account for relativistic effects, because core electrons have large kinetic energies with effective speeds a significant fraction of the speed of light. To handle many electrons, the dominant method of choice is density functional theory (DFT). While DFT simplifies calculations by relying on a single density at each point in space rather than wavefunctions dependent on a large number of coordinates (, where is the number of electrons), however, it requires an unknown functional. Nevertheless, good approximations to this unknown have been found by using theoretical constraints as well as fitting tunable parameters to experimental data.
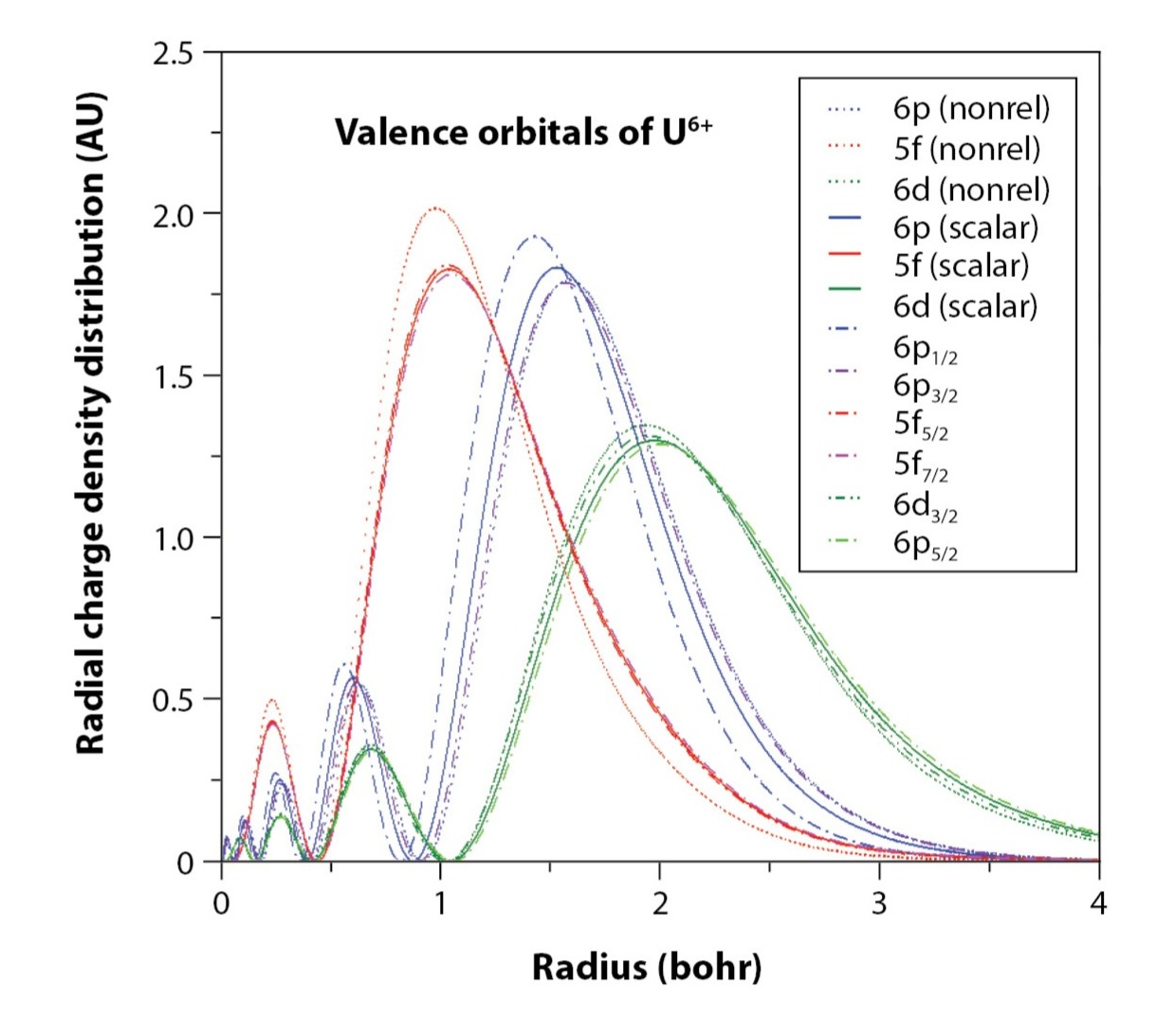
Relativistic effects can be accounted for by solving the Dirac equation instead of the Schrödinger equation, at the cost of additional complex algebra. One of the most notable effects that can be seen spectroscopically is spin-orbit coupling, terms that account for a magnetic field generated by moving electrons that in turn influence a particle’s quantum mechanical spin. As shown in Fig. 2, the radial distribution functions of valence orbitals in uranium are significantly affected by both scalar and spin-orbit corrections. This has strong implications for the strength of chemical bonding and interpretation of spectra.
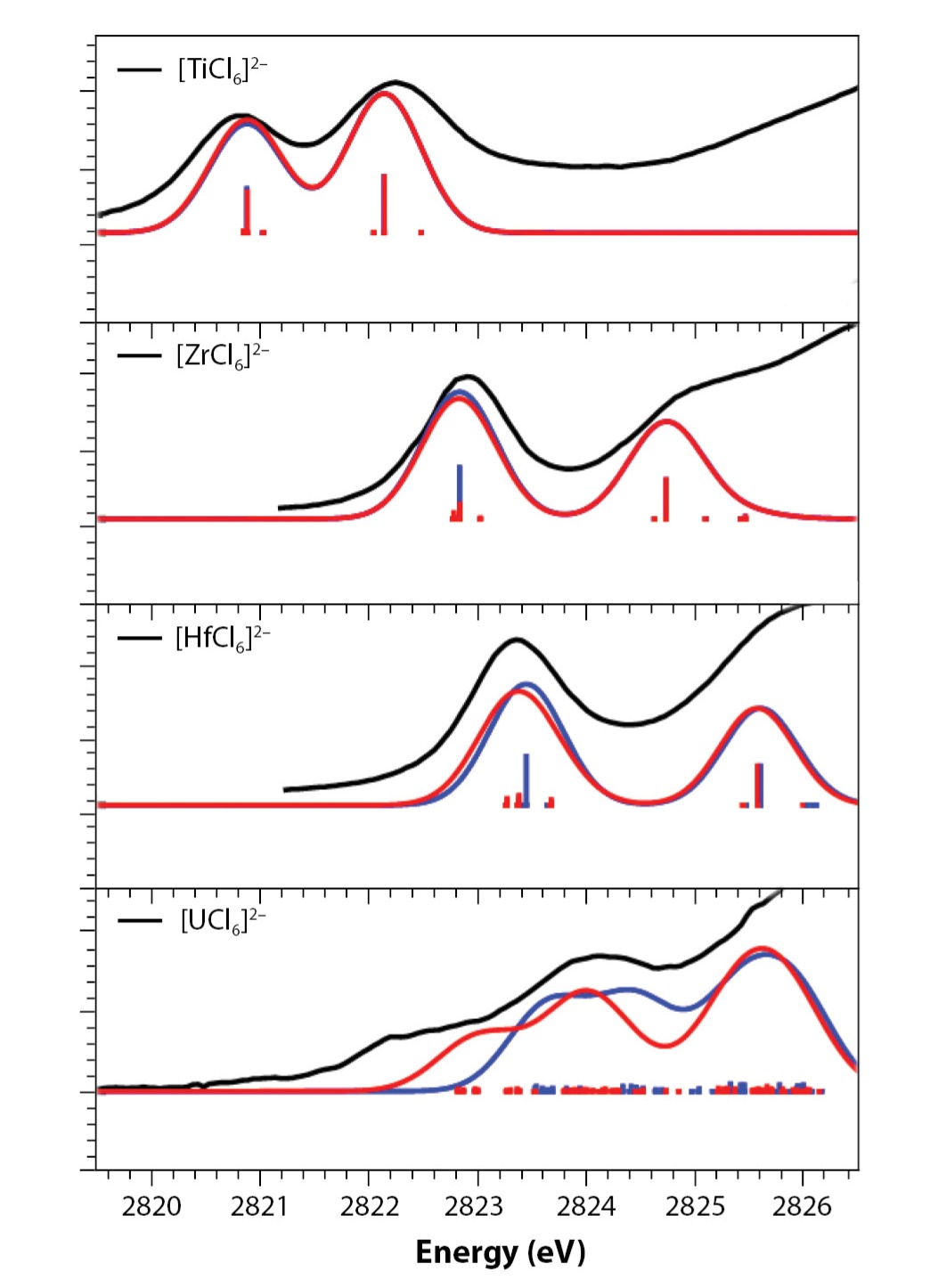
As shown in Fig. 3, while calculations that ignore relativistic corrections work reasonably well even through the 5d block, there is a noticeable disagreement between calculations that include variational relativistic effects and those that do not for the uranium chloride complex. Note that while the changes to the radial distribution functions shown previously might appear small and hard to quantify, predicting the spectra serves as a stringent test of the accuracy of a particular level of theoretical approximation due to the high sensitivity of X-ray spectra to the valence orbitals involved in chemical bonding.
Assessing the accuracy of approximations
Since variational relativistic calculations are significantly more expensive, there is a desire to use cheaper calculations when possible, either by ignoring certain terms or by using fitted potentials to simplify future calculations. Recently, we performed calculations of X-ray spectra of a variety of actinide complexes at several different levels of approximations and compared the accuracy with experimental data. Unsurprisingly, the more advanced calculations were more accurate. However, we were much more interested in the quantitative improvement. Specifically, we wanted to answer the question, “If we use this more complicated and expensive model, is it worth the effort?” Three models were compared and are shown schematically in Fig. 4. The most advanced method was known as X2CSO and included all electrons in a variational calculation with relativistic effects and spin-orbit coupling interactions. To test the value of these terms, we created a model in which these spin-orbit terms are ignored—the all-electron “scalar relativistic” calculation X2CSR model. The most inexpensive calculation was the relativistic effective core potential (RECP), a parametrized model to fit tightly bound core electrons that are not active in the problem.
For the actinide complexes studied, it was very clear that including the spin-orbit interaction provided greatly increased accuracy. A typical reduction of error with experiment was around 25–30% lower than the scalar relativistic calculation. As shown in Fig. 4, there was also a significant change to the shape and number of features. On the other hand, using the RECP method only resulted in a loss of 1–2% in accuracy compared to the all-electron X2CSR. In other words, the much cheaper RECP model could make sense for a quick estimate, but there is little reason to use the all-electron relativistic calculation without the spin-orbit terms (X2CSR), as one would spend considerably more computational effort for nearly the same result.

Emission spectroscopy and future developments
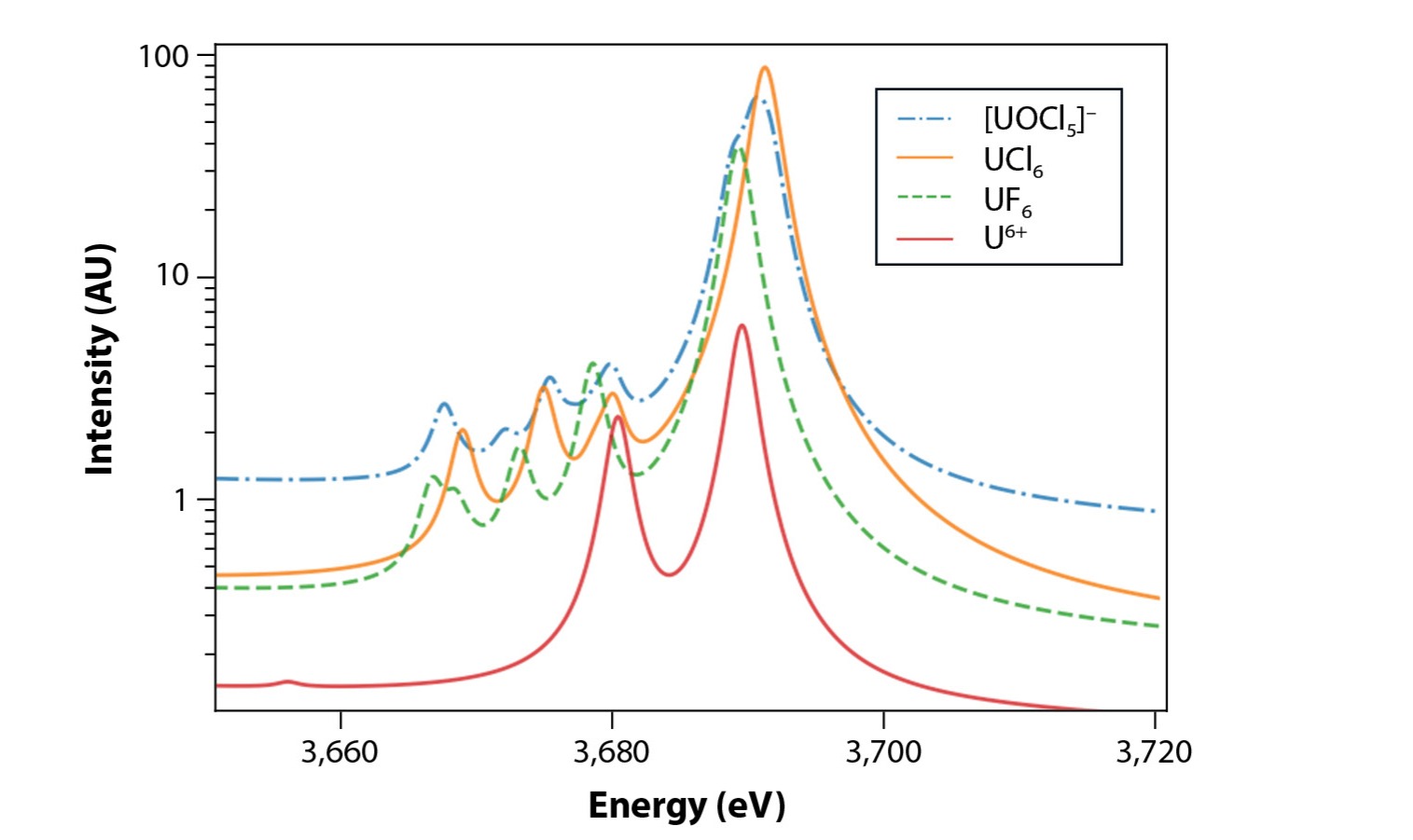
While absorption spectroscopy has been extensively used, there has been renewed interest in using its partner, emission spectroscopy, and combinations of the two, also known as resonant inelastic X-ray scattering (RIXS). These methods provide complementary information, but can have significant advantages in improving experimental data such as reduced broadening of features. New computational tools for treating these spectroscopies are underway, as most computational chemistry methods are designed to treat the ground state and not the many highly-excited states required to model emission spectroscopy. As shown in Fig. 5, current methods can capture the qualitative features, though how quantitative remains an open question.
While the calculations discussed so far represent different approximations in the choice of which physics are incorporated in the Hamiltonian, there is still a significant loss of accuracy due to the approximation of DFT and incomplete treatment of electron correlation. New computational capabilities to use the configuration interaction and coupled-cluster many-body methods with the all-electron spin-orbit Hamiltonian are being developed within the community and are expected to be essential for quantitative accuracy on experimental techniques such as X-ray emission spectroscopy and RIXS. Due to exponential scaling with the number of orbitals included for correlation, much like mentioned before, one wants to know which additional computational cost is worth paying. Systematic study will not only provide benchmark calculations for DFT results, but also aid in our understanding of which sets of orbitals are most important to chemical bonding in actinides and shed light on longstanding questions about actinyl covalency.
Summary
As computational capability has increased, the ability to accurately calculate the spectroscopy of the actinides has also improved with new methodologies and levels of approximation. While one would wish to always use the most accurate calculation, it is important to balance computational cost. Systematic studies have revealed that the expected accuracy with increased computational cost is not linear; sometimes a large jump is seen, while it can also often be the case that a much more expensive model provides very little benefit. Understanding which approximations to use for a given problem at hand remains part of the art of computational work.
About the Author

Joseph Kasper is a Scientist at Los Alamos National Laboratory within the Materials and Physical Data group (XCP-5). Previously, he worked as a postdoc with Ping Yang and Enrique Batista in the Theoretical division at Los Alamos after he obtained his PhD in chemistry at the University of Washington under the supervision of Xiaosong Li. His research interests include the electronic structure of heavy elements, computational methods for excited states, and the prediction and understanding of spectroscopy.
Acknowledgments
J.M.K. would like to acknowledge support from a postdoctoral fellowship with the Glenn T. Seaborg Institute and a Director’s Postdoctoral Fellowship at Los Alamos National Laboratory, and the Department of Energy, Office of Science (DOE-OS), Basic Energy Sciences (BES) Heavy Element Chemistry Program (HEC) at LANL, as well as collaborators on this work: Ping Yang, Enrique R. Batista, Stosh A. Kozimor, and Xiaosong Li.
Further reading:
- S. G. Minasian et al. “Determining Relative f and d Orbital Contributions to M–Cl Covalency in MCl62– (M = Ti, Zr, Hf, U) and UOCl5– Using Cl K-Edge X-ray Absorption Spectroscopy and Time-Dependent Density Functional Theory”, J. Am. Chem. Soc., 2012, 134, 12, 5586.
- J. M. Kasper et al. “Relativistic Effects in Modeling the Ligand K-Edge X-ray Absorption Near-Edge Structure of Uranium Complexes”, J. Chem. Theory Comput., 2022, 18, 4, 2171.





