
Image Caption: On the left is the electron density from a simulation for deuterium at 10 g/cc and 10 eV. On the right is the electron density for carbon at 5.7 g/cc and 8.62 eV.
Determining the fundamental properties of plasmas (e.g., equation of state, transport properties) over a wide range of temperatures and densities poses a significant challenge.
In these conditions of elevated temperature and density relative to the ambient condensed matter state, ions can be strongly coupled and electrons are at least partially degenerate, requiring a quantum treatment. These conditions have proven difficult to describe theoretically and necessitate numeric simulations.
One key approach is quantum molecular dynamics simulations. In this approach, the ions are treated classically and propagated according to Newton’s equations where the force is determined from the ions' interaction with each other and the electron density. The electron density is solved for at each ionic configuration according to the Born-Oppenheimer approximation. Density functional theory (DFT) is a formally exact method to determine the electron density of quantum electrons without reference to the many body wave equations.
In the DFT framework, the electron density is found by minimizing the free energy subject to the conservation of the total electron number. This free energy is given as a functional of the density as
F[n]=Fs[n]+FH[n]+Fxc[n]+Fei[n]
where Fs is the non-interacting free energy composed of both kinetic and entropic parts, FH is the Hartree energy or direct Coulomb interaction between the electrons, Fei is the electron-ion Coulomb interaction, and Fxc is defined as the remainder of the total free energy, which includes the quantum mechanical exchange and correlation as well as the excess kinetic and entropic terms.
Of the contributions, neither Fs nor Fxc have explicitly calculable forms, and approximations are required. In Kohn-Sham DFT, Fs may be found exactly (for a given Fxc approximation) by solving for the occupied single particle orbitals of a mean field (the Kohn-Sham) potential.
This, however, requires a matrix diagonalization to find the orbitals, which scales as the number of orbitals is cubed. To make matters worse, at finite temperature many more orbitals are fractionally occupied according to the Fermi-Dirac distribution, making the Kohn-Sham scheme intractable for very high temperatures.
In contrast, orbital-free DFT uses an approximate Fs density functional and is far less computationally expansive than the Kohn-Sham method. Both Kohn-Sham and orbital-free DFT then make use of the same orbital-free density functional approximations for Fxc.
Our work implements the orbital-free approach, and we have made significant advances in functionals for Fs and Fxc at finite temperatures that yield very high accuracy results, as detailed below. In the figure, the time for a single molecular dynamics step is plotted, showing the intractable scaling with temperature for the Kohn-Sham method. Two thresholds for the occupation numbers in the Kohn-Sham method are shown as 10−4 and 10−6.
The noninteracting free energy, Fs, is the combined noninteracting kinetic and entropic contribution from the electrons. The simplest density functional for Fs is the Thomas-Fermi approximation, which predates DFT by some 40 years. Thomas-Fermi is the local density approximation, which means that though the electron density is non-uniform, the free energy is approximated at each point in space as the energy of a uniform electron gas with density equal to the local density. Though originally developed to describe atoms at zero temperature, it is most successful in finite temperature form for hot, dense systems where in fact Thomas-Fermi represents the exact limit.
Despite this success, as temperatures lower, Thomas-Fermi quickly loses accuracy. To achieve high accuracy, one may include corrections based on not just the local density but on the local gradient of density as well. Perrot derived the first order correction at finite temperature in the formal expansion for a slowly varying density; this improves results moderately. Generalized gradient approximations have further improved results though have not been able to reproduce Kohn-Sham DFT results, which is the benchmark for orbital-free DFT where it can be calculated, across density and temperature regimes.
[Image Diagram]
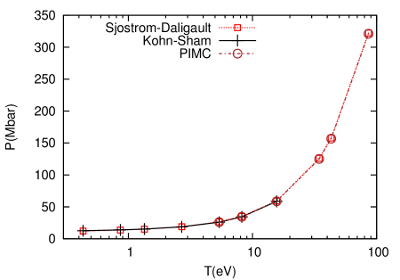
We have developed a non-local (i.e., depends on two points in space) functional by requiring a correct limit of the density-density response of the electron gas, which cannot be attained with local functionals. This has yielded highly accurate results across temperature regimes. Specifically, it is in very good agreement with Kohn-Sham methods from low to moderately high temperature, and above where Kohn-Sham calculations can be done, it is in very good agreement with highly accurate path integral Monte Carlo calculations. Results are shown here for deuterium at 4.05 g/cc. Continued work is ongoing for application to heavy element systems as well as mixtures.
The exchange-correlation free energy, Fxc, is a key contribution for which there is not an exact expression; hence, it must be approximated by a density functional that is, in general, temperature-dependent. However, while the DFT approach is increasingly used to study higher temperature systems, zero temperature exchange-correlation functionals are most commonly employed, even at significant temperature, as opposed to explicitly temperature-dependent functionals. This can result in errors of a few percent in total pressure for warm dense matter.
Recently we, with collaborators, provided fits for Fxc in the local density approximation, which were based on quantum Monte Carlo results for the uniform electron gas. These were the first fits based not on an approximate model of the electron gas but on simulation data. More recently we examined the gradient corrections to Fxc, in which we found that the temperature dependence in the gradient corrections was completely negligible. This led to a simple extension of zero temperature generalized gradient functionals to finite temperature by simply accounting for the temperature dependence in the local density term. This provides functionals that are accurate from zero to high temperature.

