What Causes Cancer?
The first large-scale inventory of cancer’s genetic fingerprints
- Craig Tyler, Editor

Why do some nonsmokers get lung cancer while some heavy smokers live full lives cancer-free? Why do most cancers develop in adults while others affect children? Why are most skin moles benign—until they’re not? In other words, what actually causes cancer?
The short answer is genetic mutations, which, in the case of cancer, generally cause one of two things to happen. Either they directly cause cells to proliferate too rapidly, or they inhibit the body’s natural mechanisms to prevent overactive proliferation. But the long answer involves which mutations, how they come about, and how they can be repaired or treated. And despite decades of effort, the long answer has largely failed to emerge. Scientists have chipped away at a towering wall of opposition, carefully extracting clues with the genetic equivalent of a fossil brush and a rock hammer. Los Alamos’s Ludmil Alexandrov, at long last, is carving it up with a light saber.
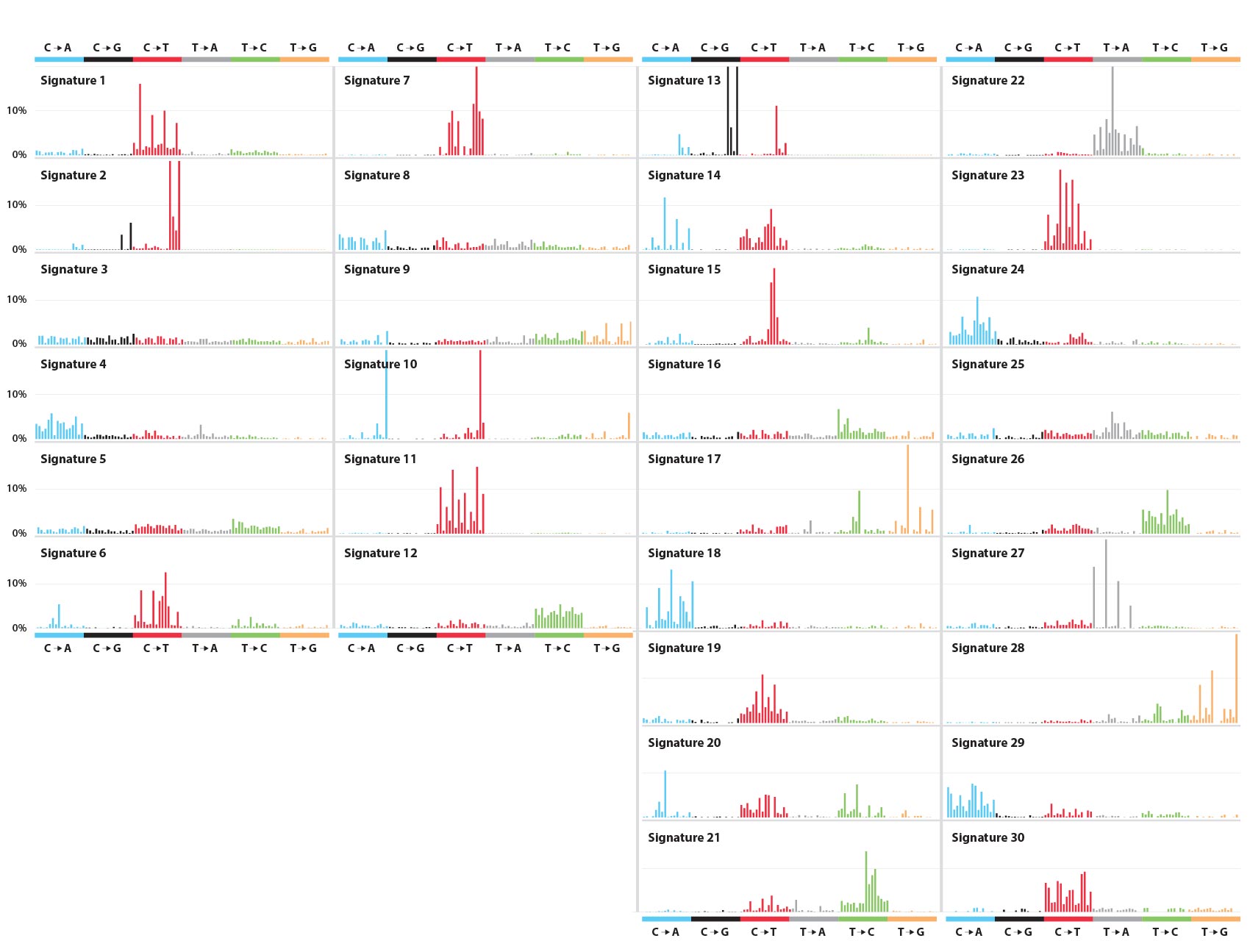
Alexandrov uses advanced supercomputers at Los Alamos to examine the full genomes of tumor cells (alongside noncancerous blood cells from the same individuals for reference) and identify mutational patterns. To date, he has analyzed the genomes from 12,023 samples spanning 40 different human cancers and identified more than 8 million distinct mutations. But mutations alone do not a cancer make, and from these 8 million mutations, he has identified 30 “mutational signatures”—recurring combinations of mutations that act like genetic fingerprints for various human cancers. Some signatures correspond to known cancer-causing defects in the genome. Others correspond to known or suspected carcinogens. Others still remain a complete mystery.
Cancer’s humble origins
“Most people think of cancer as something gone wrong,” says Alexandrov, “and that’s definitely true. But in a sense, it’s also something gone too right.” He explains that all the cells that comprise our multicellular bodies have an evolutionary history from single-celled organisms. Those organisms thrived when they were able to outcompete neighboring cells. But within a multicellular organism, that’s not so advantageous. “You don’t want an individual cell in the bladder or pancreas outcompeting all its neighbors.”
Normally, the body’s immune system prevents individual cells from getting out of control, but such immunity is imperfect. For one thing, immune cells didn’t evolve to fight modern-world cellular insults, such as tobacco, asbestos, or x-rays. For another, the immune system can become compromised by illness or immunosuppressant medications. Yet even in individuals with healthy immune systems facing naturally occurring carcinogens, immune cells, like other cells, become less effective with age. And while this, too, can be seen as a something gone right—such as when immune and other cells die or go dormant to prevent their accumulated damage from affecting the rest of the body—it still means that, with age, the immune system weakens as mutations proliferate. At some point, the problematic mutations outpace the immune system.
Ultraviolet light, tobacco, and other carcinogens are known to trigger DNA mutations that cause cancer. But which mutations and why?
Most mutations, however, are not problematic. During human cellular replication, there are typically more than 50,000 naturally occurring errors, and nearly all are automatically corrected during the process. Those that remain are bona fide mutations, yet they rarely cause any trouble. Even mutations caused by external exposures, such as chemical carcinogens or ultraviolet (UV) light, without an intense or sustained degree of exposure, rarely cause trouble. Part of the reason is that only about 1.5 percent of human DNA actually encodes for useful proteins. And even when a mutation hits that 1.5 percent, it still amounts to a very small discrepancy. It might mess up just one DNA base pair (one rung in the DNA “ladder”) out of the hundreds, thousands, or tens of thousands that make up a single gene. Such a small glitch may not actually prevent the gene, or the protein it encodes, from functioning properly. And even if the mutation were to cripple the gene, it’s only one of about 30,000 human genes. Chances are, the cell can get by without it.
The problem arises when many of these mutations combine together. A cell copies its DNA when it divides, including its acquired mutations, so all of its daughter cells have those same mutations. Thirty generations of harmless mutations down the line, say, a new mutation impairs another gene. After a couple thousand generations, the descendent cells now have a number of flaws in each of several genes. This becomes a problem if the complex pattern of accumulated mutations includes two complementary functional effects: causing excessive replication and inhibiting the genes that suppress excessive replication.
Yet such a confluence of harmful mutations still does not constitute “real” cancer. In general, when one cell undergoes excessive replication, the body’s immune system takes notice and deploys some manner of antidote. This might describe a benign skin mole, for example; a damaged cell proliferates until the body finds a way to halt its growth. To pose a danger, the mole must then acquire mutations that allow it to break through the internal cellular regulation as well as the immune system’s defenses and resume replicating uncontrollably. At this point, it’s skin cancer.
In general, a localized cancer of this sort can become life threatening in two ways. Either it grows to the point of effectively incapacitating the organ it formed within (as in liver cancer) or another mutation causes it to move beyond its organ of origin and invade other parts of the body. In the case of a mole-turned-malignant, this means moving beyond the skin and replicating uncontrollably in other organs, which are only equipped to fight their own internal cancers, not cancers of the skin. Such a metastatic cancer may start to appear all over the body, at which point the cellular proliferation proceeds unimpeded, and the patient is unlikely to survive. In fact, metastases cause 90 percent of human cancer deaths.
Tumor fingerprinting
CREDIT: Zephyris/Wikimedia Commons
While random mutations from ordinary DNA replication during one’s lifetime can cause cancer, the risk is much greater with exposure to cancer-causing agents. UV light and nuclear radiation, for example, can induce mutations in DNA by breaking its internal bonds in such a way that they reconnect incorrectly. Chemical carcinogens similarly disfigure DNA.
Cigarette smoking, for example, reliably produces the chemical carcinogen benzo[a]pyrene. A natural product of incomplete combustion—also found in coal tar, fireplace chimneys, and grilled foods—benzo[a]pyrene undergoes chemical changes in the body and subsequently bonds to the base guanine, the “G” in DNA’s “ACGT” genetic code. This distorts the double helix. When the enzymes that carry out DNA replication encounter the distortion and don’t know what to make of it, they effectively take a guess. But they guess wrong, assuming it should be a T, which pairs with A, instead of a G, which pairs with C. That’s the mutation.
Different carcinogens act differently, but the resulting DNA mutation, following replication, often takes the form of a base-pair substitution like this. Alexandrov identifies each such mutation by its incorrect genetic character substitution, as in G→T, together with the characters that come before and after for context, as in CGG→CTG. Characterized in this fashion, he identifies 96 possible mutation classes and then goes hunting for them in genomes pulled from cancerous human cells. He obtains thousands upon thousands of these genomes from the International Cancer Genome Consortium, which maintains a large and growing database of cancer genomics data, and processes them through a data-analysis pipeline he developed, running on the Laboratory’s Institutional Computing supercomputers.
“There are few places in the world that can handle the petabytes of data,” says Alexandrov. “For any given run of the analysis, a normal computer would have to chew on it for months at least. Here at Los Alamos, I can do it in a day.”
The supercomputer analysis confirmed what Alexandrov already knew, that it’s not just a single genetic character substitution that characterizes a cancer. Rather, it’s a complex blend of the 96 possible mutation classes, each with different occurrence rates. In the language of linear algebra, he creates a 96-term linear combination of mutation classes—how much class 1? how much 2? how much 96?—for each recurring pattern of mutations in his cancer genome pool. Each constitutes a mutational signature: a complicated indication of one or more types of cancer (or susceptibility to it). In turn, the cancer genomes studied—each unique to a particular cancer patient—are themselves linear combinations of mutational signatures.
You don’t want an individual cell in the bladder or pancreas outcompeting all its neighbors.
Alexandrov has identified and published 30 distinct mutational signatures to date and correlated them across the 40 different types of cancer represented in the genome pool. Signatures 1 and 5, for example, show up across the board; all 40 types of cancer show these mutational signatures. Signature 7 is consistent with classic UV-induced mutations and shows up in skin melanomas as well as oral, head, and neck cancers. Signatures 23 and 24, both of unknown origin, show up in liver cancer only.
How does someone acquire these mutational signatures? In some cases, it’s relatively easy to figure out, as with certain G→T substitutions in cigarette smokers’ DNA. Indeed, Alexandrov was able to identify different signatures particular to smokers and nonsmokers in lung cancers, as well as signatures that distinguish between smoking tobacco and chewing it. Other signatures—numbers 6, 15, 20, and 26—can be positively associated with defective DNA-mismatch repair mechanisms. And Signature 1 is apparently age-related, operating by a particular mechanism associated with cell mitosis. In other cases, there are no answers yet. Signature 5 is also likely to be age related but isn’t associated with any known mechanism. And of 13 signatures found to correlate with breast cancer, six are similarly unknown. In total, 11 of the 30 signatures have no known—or even suspected—cause.
Rather than being discouraged by so many mutational signatures of unknown cause, Alexandrov seems to value them. “You have to see the unknowns as good news,” he says. “We’re discovering completely new things about the genetic basis for cancer. This is progress. Identifying the causes and therapies will follow.”
From theory to therapy
Some cancer-causing mutations can be inherited, rather than acquired. For instance, mutations in two well-studied tumor-suppressor genes, BRCA1 and BRCA2, have long been known to associate with breast and ovarian cancers, leading some women to have their breasts or ovaries surgically removed rather than risk those tumors showing up someday. These hereditary mutations cause Signature 3. Together, the inherited and acquired mutations associated with Signature 3 constitute the combination of genetic risk, environmental exposure, and just plain bad luck that brings about actual breast and ovarian cancers.
Importantly, Alexandrov’s analysis recently revealed that Signature 3 also correlates significantly with pancreatic and gastric (stomach) cancers.
“Gastric cancer is the third-leading cause of cancer-related deaths worldwide,” Alexandrov says. “This discovery suggests a direct way to treat at least some of them.” Previous research classified patients with pancreatic cancers exhibiting a Signature 3 genetic profile (about 8 percent of them) as exceptional responders to platinum-based chemotherapy drugs. The same now seems likely to prove true for 10 percent of stomach cancers. It may also help tailor more effective treatments for about a third of breast and ovarian cancer patients—the ones matching Signature 3. The discovery also suggests that another class of drugs for treating ovarian cancer, PARP inhibitors, are likely to help with these gastric and pancreatic cancers.

Of course, treating cancer is a tricky and oftentimes discouraging business, and developing drugs that target a genetic abnormality is no exception. Several things have to go right if the drug is to make any significant difference. First, the particular genetic defect it addresses has to be utterly critical to the growth of the cancer, not just part of the mutational signature that’s along for the ride. Second, it has to be possible to create a drug that counteracts the defect in some fashion, which isn’t always the case. Third, even with an effective drug, who’s to say the temporarily thwarted cancer won’t find another defect to exploit, so it can resume its uncontrolled proliferation? This could happen either because the cancer follows a natural progression from one problem to another or because the treatment itself encourages drug resistance in the tumor cells it fails to kill.
Those caveats notwithstanding, treatments based on tumor genetics have had a real impact, reliably, if modestly, extending the lives of cancer patients. And the Signature 3 discovery is particularly promising because it implies a treatment strategy using drugs that already exist.
Yet it’s not just drug identification and development that the research may benefit. Alexandrov has shown that Signatures 1 and 5 constitute “mutational molecular clocks”—timekeepers for processes that mutate DNA on a regular schedule as a person ages. Knowledge of them may allow doctors to accurately assess the time-progression of numerous cancers, which is likely to help in selecting the optimal therapy among imperfect choices.
“The point is, we’re obtaining lots of new information about the exact mutations that cause different cancers,” says Alexandrov. “Molecular clocks and treatments for gastric and pancreatic cancers are just the beginning. We don’t yet know all the avenues for discovery and treatment that genetic fingerprinting will open up.”
Of course, to anyone suffering from cancer or from the loss of a cancer victim, such opportunities for discovery could seem like only a distant hope at best. And no wonder—at seemingly every turn, cancer has shown itself to be a hardier foe than anticipated. Yet after so many frustrating decades of trying to figure out what makes cancer tick—and more importantly, what can make it stop ticking—investigators might now be getting what they’ve needed most: a solid lead.