Making Sense of Sequences
Los Alamos bioinformatics is making it easy to interpret nature’s hereditary code.
- Rebecca McDonald, Science Writer

Out of the approximately 69 trillion cells inside a human body, more than half of them are not human. They are microbial, belonging to thousands of distinct types of bacteria, archaea, and fungi. In addition to this are countless other types of bacteria, as well as viruses, found around our living environments—our houses, our pets, our backyards, and our communities. Clues about how we cohabitate with all these organisms are buried within each one’s genetic material, or DNA. Although the vast majority of the microbes have not even been identified, much less studied, the few clues that have been deciphered foretell an enlightened future. From an understanding of the complex interactions between humans and the microbes that comprise these microbiomes may come the potential to revolutionize health and medicine.
A genome is all of the genetic information from a given organism, including genes, non-coding sequences, and mitochondrial and chloroplast genetic material. Virtual mountains of genomic data are already available thanks to advances in DNA sequencing, and it is inevitable that the mountains are only going to get bigger.
“Genomic data is being generated at a tremendous pace,” says Los Alamos bioinformaticist Patrick Chain. “In fact, it has been said that by 2025 the amount of data produced each year will outpace Twitter, YouTube, and the entire science of astronomy combined.” Chain explains that sometimes the data are used to uncover the reasons behind a disease like cancer, and sometimes they are used to trace ancestry. In other realms of biology, genomics is being used to better understand complex biological communities found in soils, lakes, oceans, or the human gut.
GenBank contains over 3 trillion bases from genomes large and small.
Bioinformatics is the interdisciplinary field that makes this analysis possible. Using DNA sequence data and bioinformatics, scientists develop knowledge about which organisms match which reference sequences; which other organisms they may be related to and in what way; how organisms function, thrive, and survive; and what relevance they have, directly or indirectly, for humankind. In order to begin answering these questions, scientists must compare unknown sequences with known ones (found in public databases, such as the primary U.S. repository named GenBank, founded at Los Alamos in 1982), and each question often requires a different approach or specialized software tool. However, regardless of the data availability, many clues remain hidden because rapid developments in sequencing technology, combined with the volume of data coming out of these machines, has created a data-analysis bottleneck. And the bottleneck is only getting tighter as the sequence data keep coming.
With this vast challenge in mind, Chain and his team at Los Alamos are making analysis easier, especially for scientists who are not bioinformatics experts. His team developed a user-friendly web interface called EDGE (Empowering the Development of Genomics Expertise) that combines openly available tools and databases to comprehensively answer any type of genomics question. And it’s working: the award-winning EDGE platform has already been deployed to at least 14 countries and is helping scientists make sense of the sequences.
The genomics era

The availability of genomic data has revolutionized how living organisms are characterized, organized, and identified—no longer by their physical traits or lifestyles but instead by their internal blueprint of DNA or RNA. As such, these data are useful for many different areas of science and medicine. For instance, sequence data can help scientists verify relationships between species based on identifying genes in common, and doctors can—although the practice is not yet widely used—determine the exact strain of flu that is making a patient sick.
To make these kinds of determinations, the sequence data must be interpreted. Although there are a multitude of shared databases and open-source algorithms available, they generally require specialized expertise, so scientists wishing to use genomics to support their research typically choose to send data to external bioinformatics experts for analysis. This approach makes reproducibility difficult because each expert may use a different tool or protocol. It also increases cost and takes valuable time. Chain’s team sought to change the paradigm by developing a way for nonexperts to use the algorithms themselves, without having to rely on external bioinformaticists’ help. This required two important steps: identifying the right tools and developing a user-friendly way to access them.
“Having a suite of tools in the same place allows you to answer several questions at once and dig deeper into the data,” says EDGE-team biologist Karen Davenport. “We’re choosing the best quality open-source tools that are not too computationally intensive and putting them together to make working with them easier.”
It’s a little like tax preparation software: instead of wading through intimidating tax code and complicated forms, the software has an attractive interface with easy-to-understand questions, the calculations are done in the background, and the software spits out a dollar amount. With EDGE, the user looks at an attractive interface where she can set question parameters, the analysis is done in the background, and the software spits out an answer—sometimes as a data visualization.
“A graphic can quickly tell you something that would take a lot more time to understand by looking at a text file or data sheet,” says Davenport.
The Human Genome printed: 109 books, 1000 pages each, 3 billion letters.
EDGE also makes analysis faster: most tasks take minutes or hours, whereas outsourcing to specialists can take days or even weeks. In addition, EDGE is open source, and it is possible to run the software on one CPU with only 16 gigabytes of memory (as on a high-end desktop computer). The development team is experimenting with cloud-based computing services as well.
EDGE 101
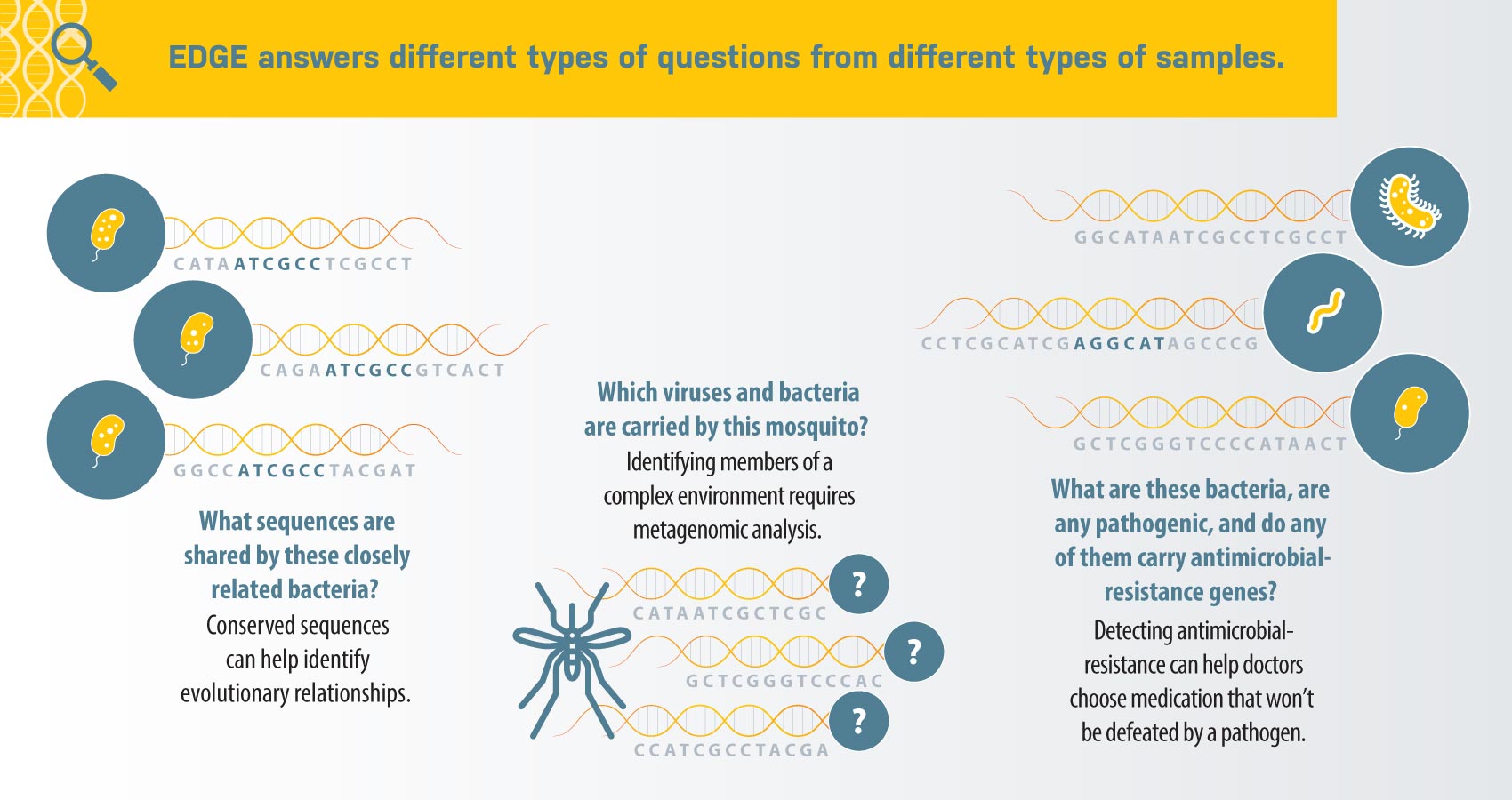
When an organism’s genome is sequenced, it is cut up into tiny pieces called “reads” that vary in length, depending on the type of sequencing machine that will be used. The machine then determines the order of the nucleic acid bases—adenine (A), thymine (T), guanine (G), and cytosine (C)—for each of the reads. Traditionally, the first role of bioinformatics is to put the pieces back together into larger contiguous sections (called contigs), which can eventually be used to reconstruct a gene (about 1000 base pairs) and then an entire genome; this is called assembly. Interpreting what the genes “say” is the next step, which involves a lot of matching against gene sequences from previously studied organisms in various databases.
Some samples of interest today, such as human-derived microbiome samples, contain more than one organism’s genome—these samples are called metagenomic. For instance, a clinical sample could contain human cells, microorganisms from the person’s microbiome, and hopefully some of whatever pathogen is making the person sick. Such diverse samples make for extremely complicated analyses because, with the exception of RNA viruses, all the reads are comprised of different arrangements of the same ATGC letters no matter what organism they came from. Therefore, it may be undesirable to do assembly first because of the number of different genomes; instead, the strategy would be to simply compare reads to various reference genomes.
“No algorithm is perfect,” says Chain. “And different perspectives can show you different things about the data.” With this in mind, EDGE was designed to have multiple options and workflows. The EDGE software uses different algorithms to answer different questions, based on the workflows chosen by users. A user might want to match genes with their function, a process called annotation, or instead the user might simply want to identify if a specific gene of interest is present.
“To determine the relatedness of a known anthrax or plague culture, we might do assembly and comparative genomics. Or to identify everything that is present in a mosquito, we would do read- or contig-based metagenomic classification,” says Chain. “Or if we are looking for antibiotic resistance genes, we would examine assembly annotations and perhaps search reads as well, using a tailored search for resistance genes.”
The EDGE platform contains over 100 published tools selected by criteria pertaining to their computational intensity and quality. The EDGE team wrote custom algorithms to make the tools work together—sometimes using the output from one tool as the input for another—and with the user interface. The team also included some of its own previously published tools, such as a database of unique signatures for microorganisms called GOTTCHA and a phylogeny module called PhaME.
On the EDGE of a breakthrough
Los Alamos postdoc Anand Kumar was trained as a veterinarian and an experimental microbiologist, so he does not have a lot of experience with writing algorithms. His current research project is to examine the disease-fighting members of the human gut microbiome. He needs to know which organisms he is dealing with and what genes they have—and the EDGE platform is helping him get results.
Specifically, Kumar wants to find out which organisms and byproducts naturally kill the bacteria Clostridiodes difficile, or C. diff, so that they can be used to treat C. diff infections. C. diff causes debilitating diarrhea and is often resistant to antibiotic treatment. When a patient has had recurring C. diff infections, he or she is often advised to undergo a fecal transplant, which involves completely cleaning out the microbiome of their intestinal tract and then re-colonizing it with a slurry of microbes from donor feces. The procedure is very effective because the microbiome of a healthy individual contains millions of beneficial bacteria, some of which secrete chemicals that can actually kill dangerous bacteria. (These types of chemicals are the origins of many current antibiotics, although there are still hundreds of unknown species and potential drugs yet to be identified.) The downside to fecal transplants, however, is they are not widely available due to FDA regulations and not without side effects and the risk of other diseases.
Using an experimental technique developed at Los Alamos, Kumar is in the process of isolating microorganisms found in successful fecal transplant samples to look for ones that show a propensity to kill C. diff bacteria. Once isolated, he can sequence them and use EDGE to analyze what he’s found—are they new species? How do they fight against C. diff bacteria? Do they have genes that are associated with potential antimicrobial activity?
Scientists are studying the human microbiome to learn which symbiotic relationships make us healthy.
Kumar’s goal is to use what he learns about these C. diff-killing microbes to create probiotic pills that people can take instead of having a fecal transplant. By including only the most effective organisms—instead of recreating the entire fecal sample in a pill—Kumar says the risks of side effects will be lower, and patients should have a better experience. Furthermore, he favors the probiotic approach over simply siphoning off the antimicrobial chemicals to be used as drugs for treatment. He explains that, although many commercially available probiotics do not tend to remain in adults’ intestines for long, the beneficial bacteria in his study originate in a healthy adult and could be different; they could colonize the new patient’s intestine permanently, leading to long-term protection.
Although EDGE is already streamlining research for scientists worldwide, one place where EDGE has the potential to make an enormous change is in the public-health sector. Antibiotics are often prescribed unnecessarily because doctors don’t have an easy and affordable way to determine exactly which bacterium or virus is making a patient sick. This misuse of antibiotics is leading to a rise in antibiotic and antimicrobial resistance.
EDGE provides tools that could help with this issue. As more medical clinics choose to purchase sequencing technology—which is already beginning and is likely to be widespread in the next few years—EDGE would make it possible for doctors and technicians to identify what pathogen is causing an illness. Furthermore, EDGE can also help determine if the culprit is resistant to certain drugs, and if so, which alternative drugs will be most effective.
Thirty years ago, the Human Genome Project prompted a revolution in sequencing technology that enabled the widespread proliferation of genomic data. It is through this flood of data that scientists have begun to fully appreciate the value of microbiomes and the symbiotic relationships humans have with microorganisms. Although the complexity of this new world view leaves scientists with more questions than answers, enlightenment is on the horizon. While some use genomics to understand what is making people sick, others are studying the microbiome to learn what symbiotic relationships make us healthy. And bioinformatics is key to making it all make sense.
More on the history of Los Alamos genome science
Three decades ago, Los Alamos scientists helped shape the future of biology by playing a foundational role in the Human Genome Project (HGP). This international project to determine the entire sequence of human DNA launched a new era of biology and medicine, but in 1986 when it was first proposed, not everyone was optimistic. In fact, many leading biologists told Congress they opposed the project, calling it “audacious” and “wasteful.” Fortunately, the vision and leadership of a few key people, including Los Alamos’s Scott Cram, Larry Deaven, and Robert Moyzis, and the late Walter Goad and George Bell, combined with the proven success of certain Lab capabilities in flow cytometry, gene library generation, and sequence database construction ultimately helped secure the $3 billion that forever expanded the reach of science.
Once the double-helix structure of DNA was resolved in the 1950s, scientists sought to determine the sequence of the chemical bases that pair together to create DNA: adenine (A), thymine (T), guanine (G), and cytosine (C), represented by a code of these four letters. The base pairs are arranged in a highly specific order that encodes all the hereditary information needed to create and maintain an organism. By the late 1970s, one could sequence about 20 base pairs in six months, and many of the sequences generated were being deposited in a publicly shared Los Alamos database called GenBank. However, as scientists began to envision sequencing the entire human genome—more than 3 billion base pairs—it became clear that doing so would require a leap in technology and strategy.
In the early 1980s, Los Alamos scientists made advances in two key areas that enabled this very leap: flow cytometry and the creation of gene libraries. Flow cytometry was invented by Los Alamos’s Mack Fulwyler in the 1960s; it works by suspending cells in liquid droplets to sort and separate them based on various properties. In 1983, Los Alamos established a National Flow Cytometry and Sorting Research Resource, through which it made numerous advances to the technology. That same year, Lab scientists also began participating in the National Laboratory Gene Library Project (in collaboration with Lawrence Livermore National Laboratory) to make libraries of flow cytometry-sorted chromosomes for distribution worldwide to labs that were researching specific genes.
In 1986, Los Alamos scientists joined colleagues and Department of Energy (DOE) leaders at a workshop in Santa Fe, New Mexico, to discuss the possibility of sequencing the entire human genome. Although there was skepticism, the success of the Library Project demonstrated that flow-sorted chromosome libraries could be used to ensure enough copies of DNA would be available for sequencing such a large genome. In 1987, the DOE funded the HGP, and in 1990, the National Institutes of Health (NIH) and many international partners joined the initiative. Each partner was assigned certain chromosomes to sequence, and throughout the project, Los Alamos and Livermore provided the critical DNA libraries. Los Alamos also took on the job of sequencing two of the 23 chromosomes: 5 and 16. In (retroactive) recognition of the value of the Lab’s seminal role in the HGP, Cram, Deaven, and Moyzis were awarded the Los Alamos Medal, the Lab’s highest honor, earlier this year.
The results of the HGP gave scientists a better understanding of genetic diseases, including cancer, but that’s not all; it also demonstrated the value of studying an entire genome. For instance, by studying the genome (instead of only particular genes) scientists have discovered that large sections previously called “junk DNA” actually encode important regulatory functions. Furthermore, the HGP showed the benefits of highly collaborative research and launched a revolution in technology that drastically reduced the cost of sequencing. With this, scientists began to sequence everything—DNA from the soil surrounding a tree root, the lining of the human gut, the handrails of the New York City subway—and it has revealed a whole new view of the world around us: one in which microorganisms vastly outnumber humans, animals, and plants. According to the NIH, the number of bases entered into GenBank from 1982 until now has doubled approximately every 18 months.
But the sequences alone do not create understanding. Quality bioinformatics is the bridge between big data and useful scientific knowledge, and this requires yet another strategic leap. That’s where EDGE comes in.
People Also Ask
In Pluribus, aliens send a genetic sequence to Earth by radio. Could DNA really be sent by radio, and could scientists actually decode something like that?
In principle, yes. A DNA sequence is just a string of characters, so once transmitted digitally, it can be received like any other radio-encoded data. The real science lies in making sense of the sequence. For more than 30 years, Los Alamos researchers have helped develop the algorithms, databases, and visualization tools that allow scientists to identify genes, trace their origins, and interpret their potential roles.
The show’s title, Pluribus is from E pluribus unum, the motto on U.S. coins meaning “out of many, one.” That’s exactly what bioinformatics does: it decodes millions (billions?) of bits of information and tries to understand how they fit together into a single organism.





